
To select the right oncology CDMO, evaluate seven criteria in this order: regulatory approvals (USFDA, MHRA, EU-GMP, ANVISA), OEB Level-5 containment certification, end-to-end oncology services, technology transfer track record, manufacturing capacity, BIOSECURE Act supply chain compliance, and quality culture. Always conduct a physical site visit before signing any contract.
Let’s be honest.
Selecting an oncology CDMO is not like choosing a vendor for office supplies. This decision can either fast-track your cancer drug to patients, or cost your company 12 to 24 months in delays, millions in wasted budgets, and in the worst case, a failed programme.
The difference between a good CDMO partnership and a bad one almost always comes down to one thing: how rigorously you evaluated the partner before you signed.
This guide gives you a clear, practical, 7-point checklist, built specifically for pharma procurement managers, VP Supply Chain leaders, and BD teams who are shortlisting oncology CDMO partners in 2026. No fluff. No theory. Just the exact questions, criteria, and red flags that separate genuinely capable oncology manufacturers from the ones who just look good in a slide deck.
Let’s get into it.
Why Choosing an On
Before we get to the checklist, you need to understand the context. Three major shifts have made oncology CDMO selection significantly more complex, and significantly more consequential, this year.
The BIOSECURE Act Has Reshuffled the Global Supply Map
The US BIOSECURE Act (H.R. 8333) came into full effect in early 2026. It prevents US federal agencies from contracting with a list of designated Chinese biotech manufacturers. The result? Global pharma companies are actively moving oncology manufacturing programmes out of China and looking for new, reliable, compliant partners.
India is the clear winner. The Indian CDMO market is valued at USD 23.3 billion in 2026, growing at a 13.2% CAGR, with a deep pool of USFDA-approved facilities ready to absorb this demand. If you are sourcing a new CDMO partner right now, you are competing for capacity at the best facilities, which makes rigorous, fast evaluation even more critical.
Oncology Drugs Are Getting More Technically Demanding
More oncology APIs today are high-potency active pharmaceutical ingredients, HPAPIs. These compounds require certified OEB Level-5 containment infrastructure to be manufactured safely. This is expensive, rare, and absolutely non-negotiable. A CDMO without it simply cannot manufacture your drug, no matter what their sales team tells you.
Every Week of Delay Is Lost Commercial Revenue
In a patent-protected commercial window, time is money in the most literal sense. A CDMO partner whose quality issues, capacity constraints, or regulatory standing creates even a six-month delay translates directly into peak-pricing revenue you will never recover. Choosing carefully at the start is always cheaper than switching mid-programme.
The 7-Point Checklist to Select an Oncology CDMO
Criterion 1, Does the CDMO Hold All the Regulatory Approvals Your Markets Need?
Regulatory approval is the first, and most important, binary filter on this checklist. If your CDMO does not hold a current, clean approval from the authority covering your target market, nothing else about them matters. You cannot sell your drug. Full stop.
Here is what each approval means in practice:
| Approval | Market Covered | Why It Matters for You |
| USFDA | United States | Required for US drug sales and all US federal contracts |
| MHRA | United Kingdom | Required post-Brexit for UK market access |
| EU-GMP | European Union (27 countries) | Required for European drug registration |
| ANVISA | Brazil / Latin America | Covers the world’s 4th largest pharma market |
| WHO-GMP | Asia, Africa, Middle East | Required for emerging market access programmes |
A multi-market approved CDMO, one holding USFDA, MHRA, EU-GMP, and ANVISA all together, means your drug can be manufactured in one location and shipped to multiple global markets without switching partners at commercial scale. That is a massive operational advantage.
Questions to ask your prospective CDMO right now:
- Can you share your current inspection certificates for each regulatory authority?
- When was your most recent FDA GMP audit and what was the finding?
- Have you ever received an FDA Warning Letter or Import Alert? If yes, show me it and show me what you fixed.
A confident, trustworthy CDMO shares all of this without hesitation. If they pause, deflect, or offer to “follow up,” that itself is a red flag worth noting in your evaluation scorecard.
Criterion 2, Does the CDMO Have Certified OEB Level-5 Containment?
This is the most technical item on the list, and one of the most important.
The vast majority of modern cancer drugs are made from high-potency active pharmaceutical ingredients, or HPAPIs. These compounds are extraordinarily potent, so much so that a microscopic amount in the air, far too small to see or smell, can cause serious and sometimes irreversible health harm to the people manufacturing them.
OEB Level-5 containment is the highest safety classification in pharmaceutical manufacturing. It means the facility can keep airborne compound exposure below 1 microgram per cubic metre of air, that is one millionth of a gram. Achieving this requires purpose-built sealed isolator machines, a fully dedicated negative-pressure air handling system, validated cleaning at nanogram-per-centimetre detection levels, and personnel wearing supplied-air respirators.
Here is what you need to know as a buyer: most CDMOs do not have this. Building a single certified OEB Level-5 manufacturing suite costs between USD 30 million and USD 80 million. Many facilities claim OEB Level-5 capability in their marketing materials, very few have the third-party certificate to back it up.
This is a binary qualification filter. Not a commercial negotiation point.
If your drug involves HPAPIs and the CDMO cannot produce a current third-party OEB Level-5 certificate from an assessor like SafeBridge Consultants, they cannot manufacture your compound safely or compliantly. Walk away.
What to ask for: The current containment certification, with the issue date, the scope of operations and compounds covered, and the containment performance benchmark data from the most recent validation run.
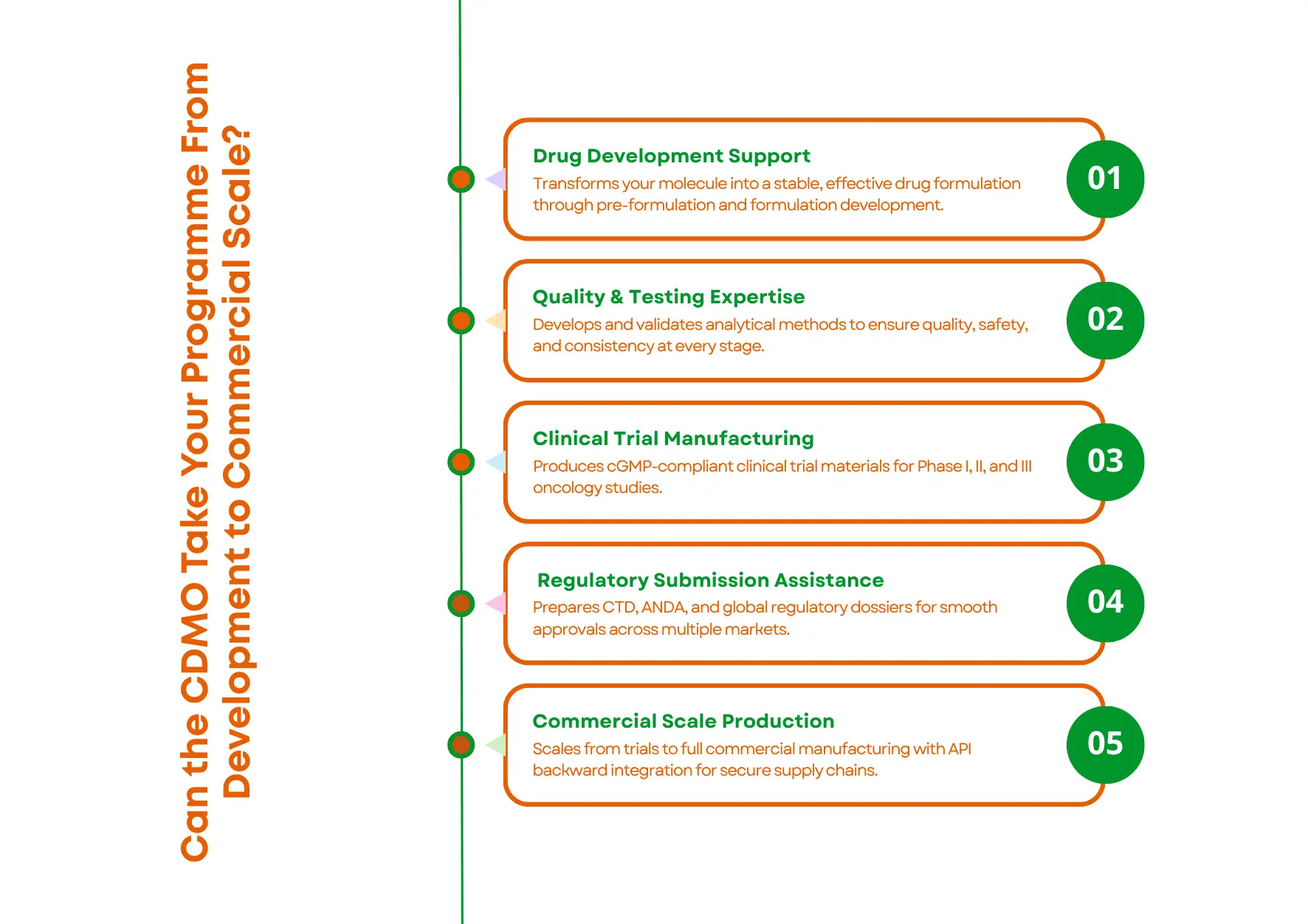
Criterion 3, Can the CDMO Take Your Programme From Development to Commercial Scale?
The strongest oncology CDMO partnerships do not just manufacture your drug, they help you develop it, prepare your regulatory submissions, and scale up with you from clinical trials through to peak commercial volume. All under one roof.
A genuine end-to-end oncology CDMO covers:
- Pre-formulation and formulation development, figuring out how to make your molecule into a stable, effective drug product
- Analytical method development and validation, creating the laboratory tests to verify quality at every stage
- Clinical trial material (CTM) manufacturing, producing GMP-grade drug product for Phase I, II, and III trials
- Regulatory dossier preparation, writing and compiling CTD, ANDA, and multi-country regulatory submissions
- Commercial-scale manufacturing, producing the volumes your markets need, with API backward integration for supply chain security
Why does this matter? When you split these stages across multiple vendors, you create handover problems. Process knowledge gets lost at every transition. Timelines slip. And when something goes wrong, which it will, no single partner owns the problem end to end.
One partner for the full journey is faster, simpler, and significantly lower risk.
Criterion 4, What Is the CDMO’s Technology Transfer Track Record?
Technology transfer, moving your drug’s manufacturing process from your lab or a previous manufacturer into the new CDMO’s facility, is statistically the most failure-prone stage of any pharma contract manufacturing partnership.
Around 50% of technology transfer delays happen because the original development team knows exactly how to make their drug work, but cannot document the process clearly enough for a new team to replicate it reliably and consistently. The result? Failed batches, root cause investigations, additional validation runs, and months of delay before the first successful GMP batch.
A CDMO with a genuine track record on technology transfer will answer all of these questions with specific numbers and documentation, not generic confidence:
- How many oncology technology transfers have you completed in the last three years?
- What is your average number of pilot runs before a successful GMP batch?
- Can you share a specific case study for a programme similar to mine in molecular complexity and dosage form?
If the answer to any of these is vague or anecdotal, ask again. A CDMO that cannot demonstrate a measurable track record on technology transfer is a CDMO whose timeline promises you should not trust.
Criterion 5, Does the CDMO Have Real Capacity Available Right Now?
Even the most technically capable oncology CDMO in India cannot help your programme if their manufacturing suites are already running at full capacity.
A facility running above 85–90% utilisation across their oncology manufacturing lines has no meaningful room for your programme, without either displacing an existing customer, delaying your start, or triggering an 18-month capital expansion that you will be waiting for.
Before committing to any CDMO partnership, ask:
- What is your current capacity utilisation rate specifically in the oncology manufacturing suites my dosage form requires?
- Can you support my programme from clinical-batch volumes all the way through to commercial scale, and what does that scale-up timeline look like?
- Do you have a confirmed capacity reservation mechanism once we sign a Letter of Intent?
Criterion 6, Is the CDMO Fully BIOSECURE Act Compliant?
If your drug programme is heading to the US market, or if any part of your company receives US federal funding, BIOSECURE Act compliance is now a mandatory due diligence check. Not a nice-to-have. A requirement.
The US BIOSECURE Act prevents US federal agencies from contracting with manufacturers that have supply chain relationships with designated Chinese biotech companies. The companies of concern list was finalised in early 2026, and even if the CDMO you are evaluating is not Chinese, their API suppliers, raw material sources, or equipment vendors may have ties that create risk for you.
Ask your prospective CDMO to provide a written declaration confirming:
- No contractual or supply chain relationships with any entity on the BIOSECURE Act concern list
- API and critical raw materials sourced entirely from compliant geographies
Indian CDMOs with vertically integrated API supply through a domestic parent company, where the API comes from a related Indian manufacturer, offer the strongest structural protection here. Their compliance is built into their supply chain design, not just declared on paper.
Criterion 7, What Does the CDMO’s Quality Culture Actually Look Like?
Documents and approvals tell you what a CDMO claims about their quality. A site visit tells you what they actually do every day when no one is watching.
Genuine quality culture shows up in the small things: how clean and organised the manufacturing floor is, how confidently the QA team talks about their systems, and whether they welcome your audit questions or answer them defensively.
Before signing, review these:
- FDA Form 483 observation history, specifically, whether the same issues recur across multiple inspection cycles
- CAPA (Corrective and Preventive Action) closure rates and timelines
- Whether electronic batch records are a genuine 21 CFR Part 11 compliant system, not scanned paper documents renamed as PDFs
- Willingness to accept unannounced audits by your QA team after the partnership begins
Rule of thumb: never sign a Master Service Agreement without a physical site visit with your own QA lead present.
Red Flags That Should End a CDMO Evaluation Immediately
Stop the evaluation, right now, if you encounter any of the following:
- The CDMO refuses or delays sharing their FDA Form 483 history or most recent audit outcome
- No dedicated oncology project manager, your programme would be handled by a generalist account manager
- OEB Level-5 capability claimed without a current, dated, third-party containment certificate
- Single-source API procurement from a tariff-exposed or sanctioned geography with no documented backup supplier
- Facility utilisation above 90% with no firm, timeline-bound capacity expansion commitment
- Electronic records that are scanned paper PDFs, not a genuine 21 CFR Part 11 digital system
- Technology transfer quoted as a fixed 3-month process regardless of your molecule’s complexity or dosage form
Any one of these is enough to disqualify a CDMO from your shortlist. Do not negotiate around them.
Why India’s Oncology CDMOs Are the Smartest Choice in 2026
India has spent 30 years building a pharmaceutical manufacturing ecosystem that now rivals the best in the world, at a fraction of Western cost. For pharma companies shortlisting oncology CDMO partners in 2026, the case for India has never been stronger.
Compared to US or European manufacturing alternatives, India-based oncology CDMOs deliver:
- Programme startup timelines up to 90% faster
- Workforce costs 70–80% lower
- Infrastructure setup costs approximately 85% lower
- Full compliance with USFDA, MHRA, EU-GMP, and ANVISA quality standards, no compromise
And the BIOSECURE Act is making this shift even more decisive. With designated Chinese CDMOs removed from US-market supply chains, USFDA-approved Indian facilities are absorbing global oncology outsourcing demand at scale. The best Indian CDMO partners, those with multi-market regulatory approvals, OEB Level-5 certified infrastructure, and vertically integrated API supply, are filling up fast.
Frequently Asked Questions, How to Select an Oncology CDMO
Q1: What is an oncology CDMO and what do they do?
An oncology CDMO, Contract Development and Manufacturing Organisation, is a company that develops and manufactures cancer drugs on behalf of pharmaceutical companies. They handle everything from early-stage formulation development through regulatory submissions and large-scale commercial manufacturing. Partnering with a CDMO lets pharma companies bring drugs to market faster, without the cost and time of building their own GMP-certified manufacturing infrastructure.
Q2: How long does it take to select and onboard an oncology CDMO?
A rigorous evaluation, from issuing RFPs through shortlisting, site visits, and contract signing, typically takes 3 to 6 months. After that, technology transfer and first GMP batch production take 12 to 18 months for oral solid dosage oncology forms. Rushing either phase is one of the leading causes of expensive mid-programme CDMO switches that set development timelines back by a year or more.
Q3: What regulatory approvals should an oncology CDMO in India hold?
For a multi-market oncology programme, your CDMO partner should hold at minimum: USFDA approval for the US market, MHRA for the UK, EU-GMP for Europe, and ANVISA for Latin America. A CDMO holding all four can supply your drug to the world’s major pharmaceutical markets from a single Indian manufacturing location, a significant commercial and logistical advantage.
Q4: What is the BIOSECURE Act and how does it affect CDMO selection in 2026?
The US BIOSECURE Act restricts US federal agencies from contracting with a list of designated Chinese biotech manufacturers. Its companies of concern list was finalised in early 2026. For any pharma company with US government contracts or federally-funded clinical programmes, selecting a CDMO with zero supply chain exposure to designated entities is now a legal compliance requirement, not a preference.
Q5: What is OEB Level-5 and why does it matter for oncology CDMO selection?
OEB Level-5 is the highest occupational safety classification in pharma manufacturing. It is required for cytotoxic oncology APIs and HPAPI compounds where even microscopic airborne quantities can harm manufacturing workers. A CDMO without a current third-party OEB Level-5 certificate cannot safely or legally manufacture your compound, regardless of what their marketing materials claim.
Q6: Does Pinnacle Life Science support end-to-end oncology CDMO programmes?
Yes. Pinnacle Life Science holds USFDA, MHRA, EU-GMP, and ANVISA approvals and supports oncology programmes from pre-formulation development all the way through commercial manufacturing of up to 3 billion tablets per year. Our OEB Level-5 certified facility in Baddi, Himachal Pradesh, is vertically integrated with API supply through Aarti Drugs Ltd., giving your programme full supply chain resilience and multi-market regulatory coverage from a single partner. Talk to our team today.

Oncology Medicine Manufacturers in India: What the Numbers Don’t Tell You
India has some genuinely strong oncology medicine manufacturers, but they’re not all built the same. Here’s what actually matters when you’re trying to find a credible partner in cancer drug manufacturing.

CDMO Companies in India: What to Know Before You Partner
India’s CDMO sector is growing fast, and for good reason. But not every partner is built the same. Here’s a practical, no-jargon guide to understanding what CDMO companies in India actually offer and how to choose wisely.

CDMO Full Form: What Is a CDMO in Pharma and How Does It Actually Work?
CDMO stands for Contract Development and Manufacturing Organization. But knowing the full form is just the starting point. Here’s what CDMOs actually do, why they matter in pharma, and how they’re different from CMOs and CROs.
